Overview
Pituitary adenomas represent a complex set of diseases. The anterior pituitary develops from a common progenitor cell into five hormone-secreting cell types, each giving rise to tumours, causing different diseases such as acromegaly and Cushing’s disease. While the majority of pituitary adenomas present as sporadic disease in adulthood, some cases present as familial disease and/or have childhood-onset. There are a few genetic syndromes where pituitary adenomas represent one of the manifestations (e.g. multiple endocrine neoplasia type-1 (MEN1) and Carney complex), while in other familial cases no other disease is associated with pituitary adenomas: these are termed familial isolated pituitary adenoma (FIPA) kindreds. While in a subset of FIPA kindreds mutations have been identified in AIP (or exceptionally rarely in GPR101), the genetic background of 80-90% of FIPA cases and around 85% of childhood-onset pituitary adenoma cases are unexplained. FIPA patients are also phenotypically heterogeneous: while patients with AIP-mutations characteristically have childhood-onset disease with predominantly GH/PRL tumours, in AIP/GPR101-negative FIPA kindreds disease onset is in middle age and tumour types are more variable. Further details you can find on https://www.qmul.ac.uk/fipa-patients/pituitary-disorders/.
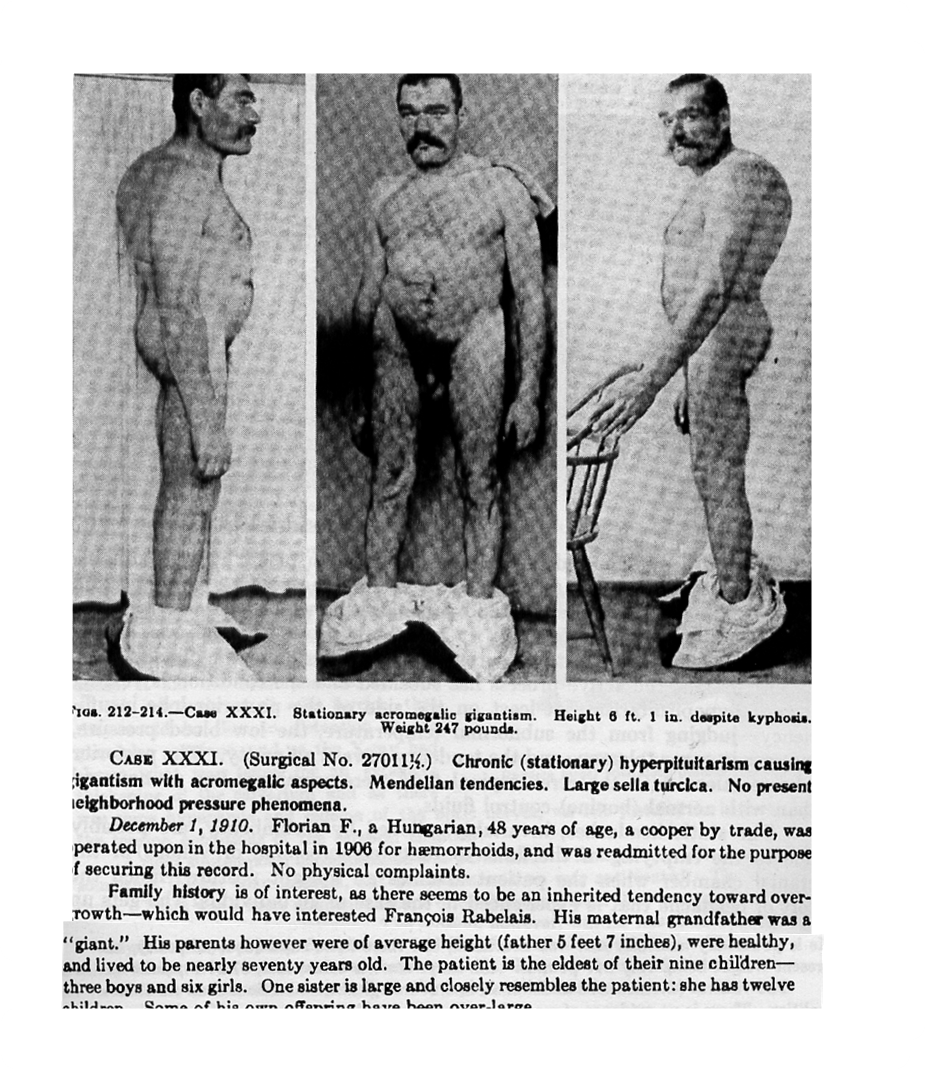
Pituitary tumours can cause severe life-long disease, especially in familial and childhood-onset cases, but earlier diagnosis would allow for treatment sooner, thus minimising suffering. We believe that patients with familial and childhood-onset disease have germline alterations predisposing to pituitary tumours.
Our two main questions are:
- What are the genetic changes causing familial or childhood-onset pituitary tumours?
- What genetic factors lead to tumours in some mutation carriers but not in others?